
And why your deep learning model keeps missing things it should catch
Let's start with a premise. You're a pathologist who's trained for a decade. I show you a small, dark, round cell on an H&E slide and ask whether it's a lymphocyte or a small tumor cell.
Sometimes you can tell instantly, sometimes you can't, and sometimes you and a colleague with identical training will give different answers and both be perfectly reasonable.
Now multiply that uncertainty by a million cells, train a deep learning model on it, and deploy it in a clinic.
This is what we've been banging our heads on for months on.
The myth of ground truth
Computer vision grew up on ImageNet, where a cat is a cat and a dog is a dog. If you squint at a few pixels maybe it gets ambiguous, but the ground truth exists because a human walked into the room and said "yes, that's a cat," and that was that.
Medical imaging borrowed the vocabulary without inheriting the certainty. We talk about ground truth labels and gold standard annotations and expert-validated datasets, using language that implies a kind of settled answer waiting to be uncovered. There isn't one, and the impact of this on deep learning has been reviewed at length but largely ignored in practice (Karimi et al., 2020).
Here's something peculiar about cell biology: most cells don't have a straight definition. Take a single cell from a tumor and ask what type of cell it is, and more often than not you're asking a question that has no clean resolution because cells exist on spectrums, sliding between states, expressing overlapping sets of markers, looking an awful lot like their neighbors. The category you want to shove them into is a box we invented, and plenty of real cells live comfortably on the seams.
So when we talk about training a model to classify immune cells or detect tumor cells or segment glands, what we're actually training it on is our best guess about what cells are, not what they are in any absolute sense.
Three ways to get "ground truth"
When you sit down to actually build a dataset, you have three options, and each one gives you different flavors of approximation.
Option one: ask a human. A pathologist looks at the cell and says what it is. This is the gold standard. But pathologists disagree with each other all the time, and published studies put inter-observer agreement on immune cell subtypes somewhere around 60 to 70% (Reynolds et al., 2023). For macrophages, which look wildly different from one case to the next, it's even worse. This isn't sloppy; it's genuine, good-faith disagreement among people who've spent entire careers staring at cells. It shows up in datasets too: the Lizard database, which relies heavily on pathologist review, reports limited inter-observer agreement on immune cell identification (Graham et al., 2021). If your labels come from one human, your dataset absorbs that human's opinions and habits and blind spots. Train ten models on ten different pathologists' labels and you'll get ten meaningfully different models.
Option two: use a molecular marker. Immunofluorescence or immunohistochemistry stains a specific protein, so cells expressing CD3 are T cells and cells expressing CD20 are B cells. Feels objective, but the process has its own persistent failure modes. Staining is physical: antibody penetration varies across the slide, signal gradients form, registration between the IF and H&E image is never quite perfect (especially at tile edges), and markers sometimes bind to things they shouldn't. A cell that is biologically a T cell can produce CD3 signal too faint to clear your threshold, while a cell that isn't a T cell can pick up enough nonspecific binding to trip the same threshold. And then there's the threshold itself; where you draw that line matters enormously, and moving it shifts your entire dataset. We ran into every one of these while building Immunocto from registered H&E/IF slides (Simard et al., 2025), and the underlying Orion dataset paper is upfront about the same limitations (Lin et al., 2023).
Option three: spatial transcriptomics. Now we're looking at gene expression, with thousands of transcripts per cell read out where the cell actually sits. Spatial transcriptomics has its own bag of issues. Transcript capture efficiency runs 10-30% on a good day, meaning most molecules in a cell are never seen. Assigning transcripts to individual cells depends on segmentation, which is itself a hard problem (Petukhov et al., 2022) (thanks, CellPose SAM!). Cell type calls from transcripts still require clustering in some latent space, which means somebody still had to pick a resolution and draw boundaries. The ground truth becomes whatever the unsupervised clustering algorithm grouped together and the human then named based on marker genes.
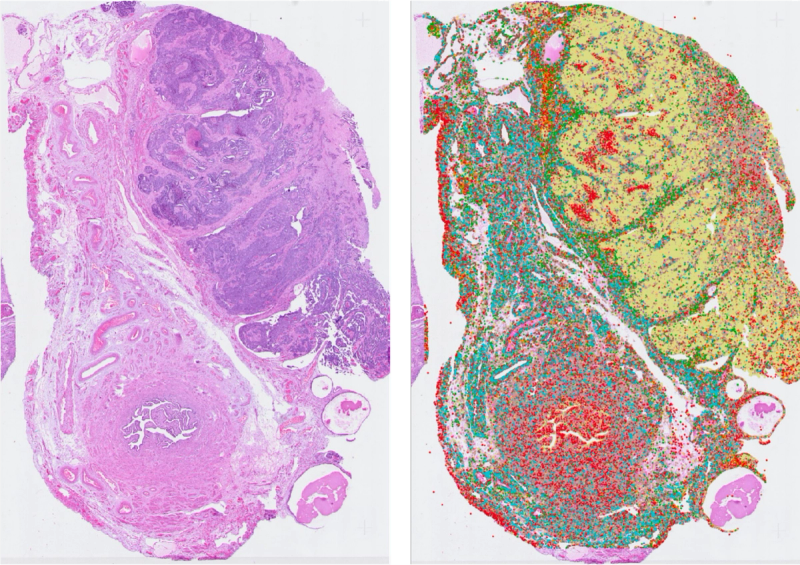
 Figure 1. How single-cell labels are generated in digital pathology. H&E paired with one of three spatial omics modalities, producing cell clusters and an annotated database to train on. Each modality brings its own systematic biases.
Figure 1. How single-cell labels are generated in digital pathology. H&E paired with one of three spatial omics modalities, producing cell clusters and an annotated database to train on. Each modality brings its own systematic biases.
The thing we're pointing at and calling ground truth keeps sliding out from under us. It's less a fixed point and more a stack of modeling decisions piled on top of each other.
The partial labeling problem
Then things go sideways in practice. All three label sources share a single failure mode: they miss cells in a systematic manner.
Humans get tired and annotate the cells they feel confident about while leaving the ambiguous ones untouched, so a dataset that claims "5,000 annotated lymphocytes" almost never has every lymphocyte in the image marked; it has the 5,000 someone was sure enough to click on. IF pipelines threshold, and cells below the cutoff get dropped from the positive class, either vanishing entirely or getting swept into a "negative" or "other" bin that now contains unlabeled positives. Spatial transcriptomics has the most severe version of this problem, since cells with too few detected transcripts can't be classified reliably and end up tagged "unknown" or defaulted into whatever cluster they land closest to in a low-quality feature space.
The result is that the cells that made it into your labeled set are not a random sample of the cells that exist. They're a biased subset.
 Figure 2. Labeled (green dots) versus unlabeled-but-similar (red arrows) cells in a single H&E tile. The model has no way of knowing the arrow-marked cells belong to the same class.
Figure 2. Labeled (green dots) versus unlabeled-but-similar (red arrows) cells in a single H&E tile. The model has no way of knowing the arrow-marked cells belong to the same class.
For a detection model (e.g. RF-DETR, YOLO), this is brutal. The model looks at a tile with ten visible lymphocytes, only three of which got labeled, and learns that the other seven are "not lymphocytes." Every time it correctly spots a lymphocyte on an unlabeled cell, it gets penalized, and the gradient pulls the model away from detecting the very thing you're trying to teach it. This is an active problem in object detection, known as the sparse annotation problem (Wu et al., 2019; Niitani et al., 2019).
For a classifier, it's sneakier but no less damaging. The model carves out a decision boundary tight around whatever subset of cells happened to get labeled, while cells outside that subset get pushed into other classes. Your validation accuracy on the labeled subset looks fine, but real-world performance is mysteriously poor. And you can't see this from inside the dataset. Every metric you check is evaluated against the same biased labels, so the model looks like it's working because it's being graded against the same flawed answer key it studied from.
Why biological label noise is different
If labels were just noisy random errors, e.g. flipped bits, deep learning would shrug it off. Big networks handle random label noise decently well, and you can train on CIFAR with 30% of labels scrambled and still pull decent accuracy because the network averages the noise away.
But biological labels aren't random. They're systematically wrong in ways that correlate with features of the data, what the label-noise literature calls feature-dependent noise, and the most damaging kind (Frénay & Verleysen, 2014).
Here's the pattern. Suppose a subset of cells are harder to stain with your IF marker, maybe because of fixation or tissue depth or some biological quirk. Those cells give weaker IF signal, your automated labeling pipeline drops them or calls them something else, and now the property that made them stain poorly correlates with "not labeled as this class" in your training data. Your network learns the correlation and learns not to call a cell a T cell if it has that property, even though the property has nothing to do with whether the cell is actually a T cell. IF staining properties, for example, becomes your GT arbitrator.
This is the kind of label noise that breaks models in the wild. It doesn't show up as poor validation accuracy; it shows up as models that hum along beautifully on the test set and then stumble in clinical deployment, because the test set carries the same bias as the training set. The bias stays invisible until someone holds you to a standard your labels never captured.
The diffuse morphology problem
Some cell classes announce themselves clearly. A plasma cell has that distinctive clock-face nucleus, a mitotic figure has its characteristic spindle, and a pathologist can point and say that one with most colleagues nodding along.
Other cell classes are smeared across a much wider range. Their morphology covers a lot of ground and they look like many things, so you can see a hundred examples and only half of them share any obvious visual thread with the others.
Macrophages are the classic case. A macrophage can be a tidy round cell with a kidney-shaped nucleus, or a giant foamy thing stuffed with phagocytosed debris, or a cell mid-mitosis, or something tucked into tissue looking almost identical to a fibroblast. What connects them biologically doesn't translate cleanly into anything visual, a problem compounded by the fact that macrophages tend to intermix with other cell types rather than forming tight clusters (Hickey et al., 2023).
For diffuse classes, the label noise compounds because not only is your labeling signal shaky, but the class you're trying to pin down is itself fuzzy. Two pathologists looking at a borderline cell have no shared answer to land on, and two IF markers give different positivity rates for "macrophage" because they're capturing overlapping but not identical populations.
This is where models hit a wall. Lymphocytes, for all their variability, still hang together as a morphological class, but macrophages don't. Train the same architecture on the same pipeline and you might get 70%+ F1 on lymphocytes and 40% on macrophages, and it's not because the model is suddenly worse; it's because "macrophage" as a label is mushier than "lymphocyte" at every level: human judgment, molecular definition, visual signature.
The multiplex stain-from-H&E pipeline
All this feeds into a research direction that's picked up considerable steam in recent years: predicting stains or IF from H&E (Latonen et al., 2024).
The pitch is elegant. IF is expensive, destructive, and not part of routine clinical workflow, while H&E is everywhere. If you could train a model to predict molecular signal (CD3, CD8, CD68) straight from H&E, you'd unlock molecular characterization of tissue from a routine stain with no extra cost and no extra slides.
And to be fair, papers do report correlations. On a good day, for certain markers and well-controlled datasets, you'll see Pearson's r around 0.6 or 0.7 between predicted IF and ground truth IF on held-out slides (Burlingame et al., 2020; Bian et al., 2024). The numbers look real and the visual overlays can be striking.
The first problem: that's only for markers where the underlying biology does produce some visible morphological signature. There are plenty of molecular signals that leave no consistent trace in H&E morphology at all. A cell can upregulate a receptor or secrete a cytokine without changing its shape or its staining pattern in any way that a model could possibly latch onto. For those markers, the model has nothing to work with because the thing you're asking it to predict is fundamentally invisible in the input.
And even for the markers where you can hit that 0.6 or 0.7 ceiling, the "ground truth" IF image is the same IF signal that drops out for weakly staining cells, that suffers from registration error, that drifts in gradients across the slide, that thresholds differently under different settings. When your model predicts that signal with reasonable accuracy, you're congratulating it for reproducing the measurement, not necessarily the biology underneath. If your model underpredicts IF signal in a region where the actual IF was also weak because of poor antibody penetration, your correlation ticks up, and the model learns the staining artifacts right alongside whatever morphological cues it managed to find. From the outside, you can't tell them apart.
The final issue: even if the model nailed IF prediction perfectly for a given marker, it would still inherit every limitation of IF as a label source. A T cell with weak CD3 staining in reality will have weak predicted CD3 from the model, so you haven't escaped the label noise; you've just shifted it from training time to inference time.
This isn't to say IF-from-H&E is pointless. For markers that do have some morphological correlate, and for applications where you care more about spatial patterns and rough density estimates than single-cell precision, it can be genuinely useful. But it's not perfect, and the model predicts a noisy measurement of a noisy measurement of biology we can't observe directly, and it only works at all when that biology bothers to show up in the pixels. Calling its output "predicted CD3" makes it sound like we've gained a molecular lens when what we really have is a regression onto a slightly flawed target, with a ceiling that varies wildly depending on how much the target actually reveals itself in an H&E stain and the reproducibility of the training data.
The bigger picture
Deep learning's loudest successes have come in domains with cheap, high-agreement labels. Is this a cat? Is this English? Did the user click? Even when labels are noisy, there's usually a latent truth and the noise is mostly about measurement error.
Cell biology is different because the latent truth is itself a modeling choice. When we say a cell is a T cell, we're compressing a continuous biological reality into a discrete bin someone found useful. The category isn't wrong, but it isn't absolute truth.
Partial labeling makes it worse. Imperfect labels across every cell would be manageable, but perfect labels on a biased subset are a trap. You can't see the problem from inside the dataset and your metrics hum along, then you deploy and a chunk of cells your model should have caught slip through, and you can't figure out why.
If you're training models in this space, you'll eventually hit the ceiling of your labels. Performance will flatten and a bigger network can't fix it.
But is this actually a problem in digital pathology? Probably not uniformly. For coarse tissue-level signal (infiltration density, prognostic stratification, spatial patterning) imperfect labels have already proven good enough (Shen et al., 2025). For anything that needs single-cell precision, rare-class detection, or a quantitative biomarker claim, the ceiling your labels impose becomes the ceiling your model will hit. And to be completely fair, with immune cells and tumour cell density, we don't know which side of this limit we are in.
